الثلاسيميا بشكل مفصل
الثلاسيميا’ α-thalassemia
مرض ينتمي إلى عائلة من الأمراض الوراثية التي تؤثر في الدم البشري المعروف باسم اضطرابات الهيموجلوبين أو اعتلال الهيموجلوبين.
اضطرابات الهيموجلوبين أو متغيراته هي مجموعة من الاضطرابات تؤثر على مستوى الهيموجلوبين، وهو بروتين يحتوي على عنصر الحديد الذي تحمله كريات الدم الحمراء، ومن هنا جاءت التسمية باضطرابات الهيموجلوبين أو اعتلالاته أو متغيراته.
اضطرابات الهيموجلوبين، بما يشمل مرض ’الثلاسيميا’ تعد مصدر قلق دولي.وتشير الإحصائيات العالمية أن ما يقارب 7% من إجمالي السكان بالعالم حاملون لأمراض الاضطرابات في الهيموجلوبين. ونحو 500-300 ألف طفل يولدون مصابين بأمراض الاضطراب في الهيموجلوبين الكبرى سنويا.
إلا أن التقدم العلمي في مجال الرعاية السريرية لمرضى ’الثلاسيميا’ قد ساهم في تحويله من مرض يهدد الطفولة حتى وصل الأمر لاعتباره مرضا مزمنا، لأن يكون حاليا مرضا يسهل التحكم في حدته حتى بلغ معدل تعايش المرضى في تزايد مستمر ما ساعد في تطوير حياتهم.
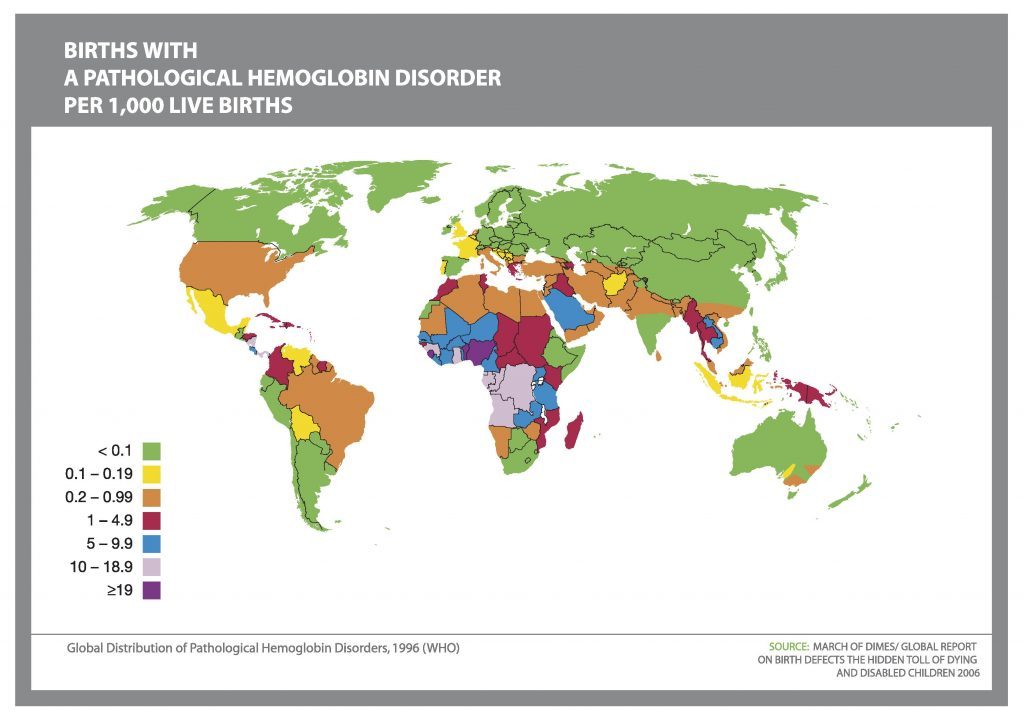
ألفا ثلاسيميا Alpha Thalassemia
THE MAJOR ALPHA (α-) THALASSAEMIAS ARE:
مرض HbH
α-thalassemia Hydrops Foetalis
(= Hb Bart’s Hydrops Foetalis)
تحدث العديد من الأمراض في الإنسان بسبب تشوهات في الدم ويتم تصنيفها وفقًا لعنصر الدم المتأثر: أمراض الخلايا البيضاء وأمراض الصفائح الدموية وأمراض الخلايا الحمراء.
اضطرابات الهيموغلوبين أو اعتلال الهيموغلوبين هي مجموعة من الحالات التي تؤثر على دم الإنسان – وعلى وجه التحديد مادة أو بروتين مهم يسمى الهيموجلوبين الموجود في خلايا الدم الحمراء ، ومن هنا تأتي اضطرابات الهيموجلوبين أو اعتلال الهيموغلوبين.
الهيموجلوبين هو بروتين يتكون من أجزاء أو سلاسل وتسمى سلالسل ألفا (α) أو سلاسل بيتا (β) والتي تنتج بدورها من جينات ألفا غلوبين وجينات بيتا غلوبين على التوالي. ومن هنا تنقسم الأمراض الناجمة عن شذوذ الهيموجلوبين سواء فيما يتعلق بإنتاجه أو بهيكله إلى أمراض سلسلة ألفا (أو جينات α-globin) ، مثل α-thalassemia ، وأمراض سلسلة β-(-globin ، مثل β-thalassemia الكبرى أو العظمى ومرض الخلايا المنجلية. تم العثور على هذه الجينات على الكروموسومات 16 و 11 على التوالي ، وإنتاج كميات متساوية من سلاسل ألفا و سلاسل بيتا على التوالي والتي تتطابق مع α2β2 لإنتاج هيموجلوبين البالغين.
بيتا ثلاسيميا β-thalassaemia
ما هي بيتا ثلاسيميا ؟
هنا سنتحدث عن متلازمات الثلاسيميا بيتا (β-) التي يكون فيها ثلاسيميا بيتا الكبرى ، المعروف أيضا باسم الأنيميا المتوسطة أو أنيميا كولي ، وهي الأكثر شدة سريريا.
أنواع بيتا ثلاسيميا (β-) هي:
β-thalassaemia major بيتا ثلاسيميا العظمى
β-thalassaemia intermediate بيتا ثلاسيميا الوسطى
HbE/β-thalassaemia بيتا ثلاسيميا HbE
Other rare thalassaemias ثلاسيميا نادرة أخرى
تحدث العديد من الأمراض في الإنسان بسبب تشوهات في الدم ويتم تصنيفها وفقًا لعنصر الدم المتأثر: أمراض الخلايا البيضاء وأمراض الصفائح الدموية وأمراض الخلايا الحمراء.
اضطرابات الهيموغلوبين أو اعتلال الهيموغلوبين هي مجموعة من الحالات التي تؤثر على دم الإنسان – وعلى وجه التحديد مادة أو بروتين مهم يسمى الهيموجلوبين الموجود في خلايا الدم الحمراء ، ومن هنا تأتي اضطرابات الهيموجلوبين أو اعتلال الهيموغلوبين.
الهيموجلوبين هو بروتين يتكون من أجزاء أو سلاسل ألفا (ألفا) وبيتا (بيتا) أو سلاسل والتي تنتج بدورها من جينات ألفا غلوبين وجينات بيتا غلوبين على التوالي. ومن هنا تنقسم الأمراض الناجمة عن شذوذ الهيموجلوبين سواء فيما يتعلق بإنتاجه أو بنيته إلى أمراض سلسلة ألفا (أو جينات α-globin) وأمراض السلسلة β-globin. تم العثور على هذه الجينات على الكروموسومات 16 و 11 على التوالي.
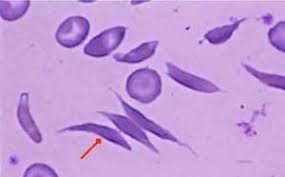
لا يستطيع مرضى الثلاسيميا الكبرى ، وهي أشد أشكال الثلاسيميا ، أن يجعلوا الهيموجلوبين البالغ الطبيعي ، الذي يتكون من أعداد متساوية من سلاسل ألفا وبيتا ، ونتيجة لذلك لا يمكن أن ينتج خلايا الدم الحمراء الطبيعية (RBCs). في هؤلاء الأفراد ، أي أولئك الذين لديهم ثلاسيميا عظمى، تحتوي كل خلية دم حمراء على كمية أقل بكثير من الهيموجلوبين ، لأن جينات β-globin لا تعمل أو تعمل بشكل غير صحيح وبالتالي لا تنتج أو تنتج كميات صغيرة جدا من السلاسل β. وبالتالي هناك خلايا حمراء أقل بكثير من المدى الطبيعي. هذا يسبب فقر الدم ، وهو شديد جداً في هؤلاء المرضى.
علاج الثلاسيميا
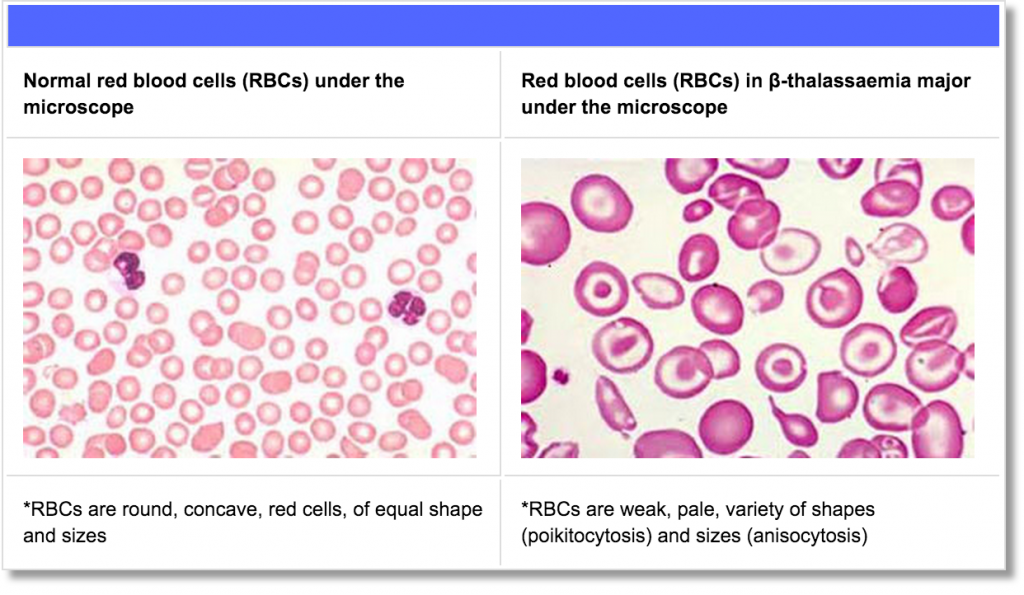
في الوقت الحاضر ، النهج الوحيد المتاح لعلاج الثلاسيميا هو زرع نخاع العظم ، وفقا للمتطلبات المحددة التي يجب على كل من المتبرع والمريض الوفاء بها. ويجري دراسة العلاج الجيني كعلاج محتمل لبعض المرضى.
زرع نخاع العظم
زرع نخاع العظم (BMT) أو زرع الخلايا الجذعية المكونة للدم (HSCT) هو إجراء طبي يتم فيه نقل الخلايا الجذعية (وهي أنواع خاصة من خلايا الدم) من فرد سليم (المتبرع) إلى دم شخص مصاب بمرض دم مثل الثلاسيميا β (المتلقي).
الخلايا الجذعية هي الخلايا الأولية المنتجة في النخاع (الأنسجة الرخوة) لبعض العظام الكبيرة في الجسم. تتطور الخلايا الجذعية تدريجيًا إلى خلايا عالية التخصص ، على سبيل المثال ، خلايا الدم الحمراء (RBCs). هذه الخلايا تحتوي على الهيموغلوبين ، تتطور من نضوج الخلايا الجذعية ولديها القدرة الخاصة على حمل الأكسجين عبر أعضاء الجسم والأنسجة.
يمكن أخذ الخلايا الجذعية من النخاع العظمي للتبرع (bone marrow donation)؛ أو من مجرى الدم (peripheral donation) ؛ أو من دم الحبل السري من المولود الجديد (through the umbilical cord). إذا نجحت ، يمكن أن تقدم BMT / HSCT علاجاً كاملاً ودائماً لمرضى الثلاسيميا بيتا بما في ذلك عدم الحاجة لعمليات نقل الدم.
الرعاية الصحية والنفسية لمرضى الثلاسيميا
خلايا الدم الحمراء (RBC) للأشخاص الذين يعانون من ثلاسيميا بيتا كبيرة (صغيرة الحجم) وغير ناضجة عند إطلاقها في الدورة الدموية، ليس لديهم التركيز الصحيح للهيموجلوبين (هيموكرومي) ، فهي هشة وغير فعالة في نقل الأوكسجين في جميع أنحاء الجسم لذلك فهي تتضرر وبالتالي فهي مستهدفة لتدمير الخلايا الشبكية. عندما يحدث هذا يتم استخراج الحديد التي تحتوي عليها وتخزينها في الكبد لإعادة استخدامها في صنع كرات الدم الحمراء الجديدة.
يبقى نقل الدم الطويل مدى الحياة هو العلاج الوحيد المتاح، لذلك يعتمد الأفراد على الإدارة الطبية طويلة المدى ويتطلبون نقل الدم على فترات تتراوح من أربعة إلى ثمانية أسابيع تقريبًا.
تجدر الإشارة إلى أن الأفراد لا يزالون قادرين على امتصاص الحديد من نظامهم الغذائي، هذا جنبا إلى جنب مع الحديد المنطلق من كرات الدم الحمراء المدمرة قبل الأوان، والحديد في الدم الذي يتم نقله يزيد من كمية الحديد في الجسم ويؤدي إلى تراكم الحديد الزائد الشديد والمزمن.. هذا هو السبب الرئيسي لتلف الأعضاء الرئيسية في القلب والكبد والبنكرياس والكلى على وجه الخصوص.
من أجل تسهيل إفراز الحديد الزائد من الجسم وتصحيح الأفراد الذين يعانون منه ويحتاجون إلى العلاج بإستخلاب الحديد. يتم ذلك عن طريق الحقن الذاتي (في حالة الأطفال يتم ذلك من قبل الوالدين) لعامل مخلب يسمى( desferrioxamine, Desferal). يدار هذا باستخدام الوخز المستمر تحت الجلد عن طريق جهاز لضخ الديسفرال، ويتم إعطاؤه في المتوسط لمدة خمس ليال في الأسبوع ، يعمل لمدة 8 ساعات تقريبًا لكل جلسة علاج.
تعتمد فعالية إزالة هذا المعدن الثقيل( الحديد) على استعداد الفرد وقدرته على الالتزام بالمعالجة الموصوفة. بعد سنوات عديدة من إجراء أربع عمليات نقل دم أسبوعية، خمس ليال في الأسبوع، وعلاج مساعد آخر يجعل العيش مع الثلاسيميا مرهقاً كبيراً، وقد يفشل الأفراد في الحفاظ على علاجهم، وبالتالي يخاطرون بتطور المضاعفات القاتلة خاصةً زيادة حمولة الحديد. كما هو الحال مع ظروف الحياة المزمنة الأخرى، فإن الإدارة النفسية لمجموعة المرضى هذه ضرورية ويجب أن تكون جزءاً من خطة الرعاية الشاملة.
لا يعتبر خالب Deferiprone الفموي فعالاً في إزالة الحديد الزائد مثل desferrioxamine ولكنه أفضل من لا شيء بالنسبة لأولئك المرضى الذين يعانون من صعوبة في الامتثال لاستخدام علاج إزالة الحديد تحت الجلد. وقد ثبت أن الخالب Exjade الفموي أكثر فعالية من Deferiprone لكن تكلفته باهظة إلى حد ما. وقد أصبح الدواء أكثر استخدامًا في بعض البلدان.
تشمل العلاجات الأخرى لمجموعة المرضى هذه استخدام المضادات الحيوية بشكل يومي واستخدام فيتامين C لمساعدة Desferal في دورها كعامل مخلب. ويعطى المرضى غير المنقولين أيضا حمض الفوليك 5 ملجم / أسبوعياً
المراقبة عن كثب لوظائف القلب والكبد والغدد الصماء أمر حيوي من أجل تحديد المضاعفات الوشيكة بهدف منعها إذا أمكن أو إدارتها إذا لم يكن من الممكن السيطرة عليها بالكامل. وفي سن البلوغ لابد من مراقبة هذه الوظائف وإدارتها عن كثب.