All about Thalassemia
About Thalassaemia
Thalassaemia belongs to a family of genetic conditions affecting the human blood, known as Haemoglobin disorders or Haemoglobinopathies. Haemoglobin disorders are a group of conditions affecting haemoglobin, an important substance or protein of the human blood contained in the red blood cells, hence the name haemoglobin disorders or haemoglobinopathies.
Haemoglobin disorders, including thalassaemia are an international concern. It is estimated that 7% of the world population is a carrier of a severe haemoglobin disorder and 300-500.000 children are born with a severe haemoglobin disorder each year. Research advances in the clinical care of thalassaemia have managed to transform thalassaemia from a fatal disease of childhood that it once was into a chronic, yet well-managed disease, increasing patients’ survival rate and improving their quality of life.
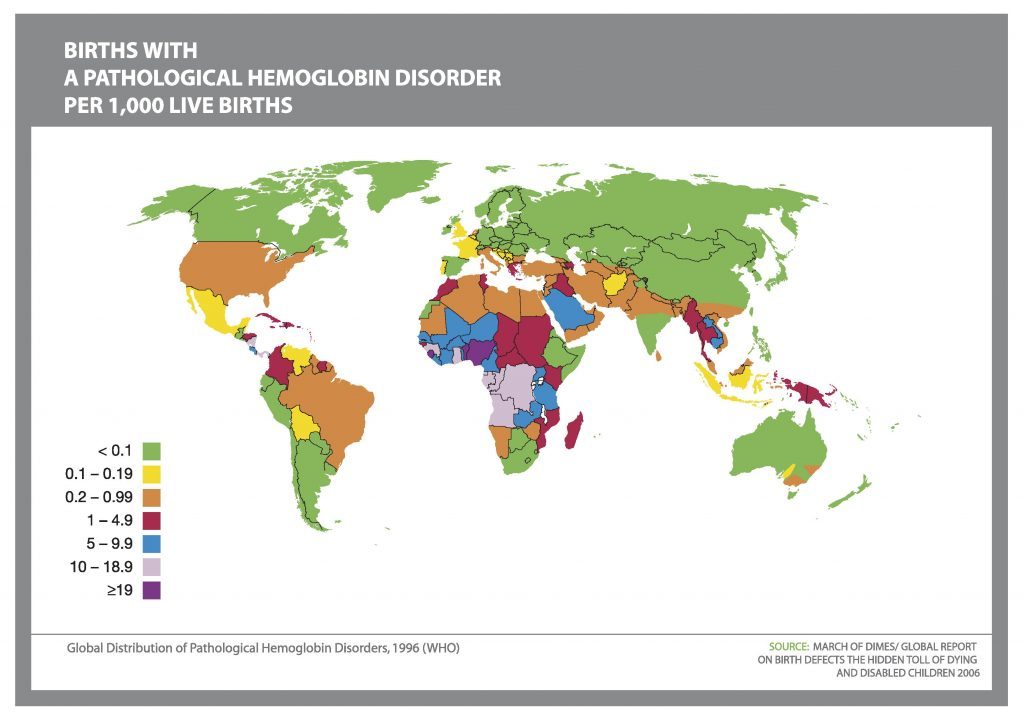
Alpha thalassaemia
Here we will speech about alpha (α-) thalassaemia.
THE MAJOR ALPHA (α-) THALASSAEMIAS ARE:
HbH disease
α-thalassaemia Hydrops Foetalis
(=Hb Bart’s Hydrops Foetalis)
Many diseases in humans are caused by abnormalities in the blood and these are categorized according to the component of the blood which is affected: white cell diseases, platelet diseases and red cell diseases.
Haemoglobin disorders or haemoglobinopathies are a group of conditions affecting human blood – more specifically an important substance or protein called haemoglobin contained in the red blood cells, hence the name haemoglobin disorders or haemoglobinopathies.
Haemoglobin is a protein that consists of the alpha (α) and beta (β) parts or chains and which are in turn produced by the α-globin genes and β-globin genes respectively. Hence the diseases caused by haemoglobin abnormality either with regards to its production or its structure are divided into α-chain diseases (or α-globin gene) diseases, such as α-thalassaemia, and β-chain (β-globin gene) diseases, such as β-thalassaemia major and sickle cell disease. These genes are found on chromosomes 16 and 11 respectively, producing equal amounts of α and β chains respectively which match together to α2β2 to produce the normal adult haemoglobin (HbA, α2β2).
Beta Thalassaemia
What is beta thalassaemia
This section is dedicated to beta (β-) thalassaemia syndromes of which β-thalassaemia major, also known as Mediterranean anaemia or Cooley’s Anaemia, is the clinically most severe one.
THE MAJOR BETA (β-) THALASSAEMIA SYNDROMES ARE:
β-thalassaemia major
β-thalassaemia intermedia
HbE/β-thalassaemia
Other rare thalassaemias
Many diseases in humans are caused by abnormalities in the blood and these are categorized according to the component of the blood which is affected: white cell diseases, platelet diseases and red cell diseases.
Haemoglobin disorders or haemoglobinopathies are a group of conditions affecting human blood – more specifically an important substance or protein called haemoglobin contained in the red blood cells, hence the name haemoglobin disorders or haemoglobinopathies.
Haemoglobin is a protein that consists of the alpha (α) and beta (β) parts or chains and which are in turn produced by the α-globin genes and β-globin genes respectively. Hence the diseases caused by haemoglobin abnormality either with regards to its production or its structure are divided into α-chain diseases (or α-globin gene) diseases and β-chain (β-globin gene) diseases. These genes are found on chromosomes 16 and 11 respectively.
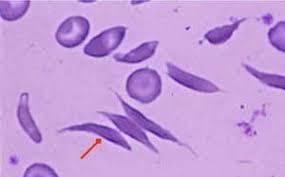
Patients with β-thalassaemia major, the most severe form of thalassaemia, cannot make normal adult haemoglobin, which is made up of equal numbers of α- and β-chains, and as a consequence cannot produce normal red blood cells (RBCs). In these individuals, i.e. those with β-thalassaemia major, each red blood cell contains much less haemoglobin, because the β-globin genes are not working or functioning properly and thus do not or produce very small amounts of β-chains. Consequently, there are far fewer red cells than the normal range. This causes anaemia, which is severe in these patients, as shown in the figure below on the right hand side.
Thalassaemia cure
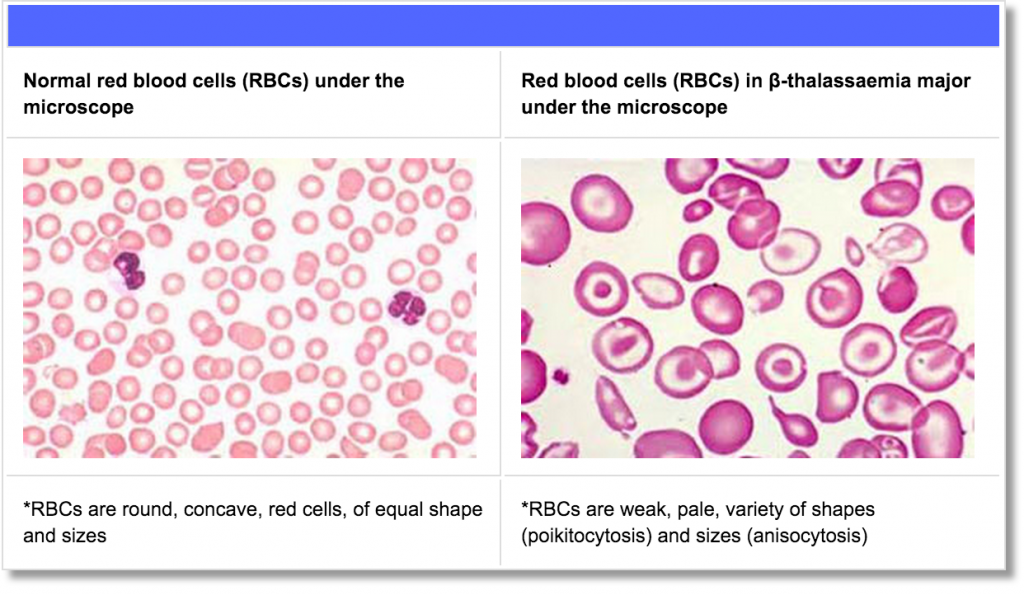
At present, the only available approach to curing thalassaemia is bone marrow transplantation, following specific requirements that both the donor and patient must fulfil. Gene therapy is being studied as a potential cure for some patients.
Bone Marrow Transplantation
Bone Marrow Transplantation (BMT) or Haematopoietic Stem Cell Transplantation (HSCT) is a medical procedure during which stem cells (a special types of blood cells) are transferred from a healthy individual (the donor) into the blood of an individual with a blood disease such as β-thalassaemia (the recipient).
Stem cells are starter cells produced in the marrow (soft tissue) of some large bones of the body. Stem cells progressively develop into highly specialized cells e.g., Red Blood Cells (RBCs). RBCs, containing Hb, develop from the maturation of stem cells and have the specialized ability to carry oxygen across the body organs and tissues.
Stem cells can be taken from the donor’s bone marrow (bone marrow donation); bloodstream (peripheral donation); or cord blood from a newborn baby (through the umbilical cord). If successful, BMT/HSCT can offer a complete and permanent cure to β-thalassaemia patients, including no need for blood transfusions.
Thalassaemia Clinical Management
The Red Blood Cells (RBC) of people with beta Thalassaemia major are small (microcytic) and immature when released into circulation, they do not have the right concentration of haemoglobin (hypochromic), they are fragile and inefficient at carrying oxygen around the body, they become damaged and are therefore, targeted for destruction by the reticuloendothelial cells. When this occurs the iron they contain is extracted and stored in the liver for re-use in making new RBCs.
Life long blood transfusion remain the only treatment available therefore individuals are dependent on long term medical management and require transfusion at four to eight week intervals approximately.
It should be noted that individuals are still able to absorb iron from their diet, this combined with the iron released from prematurely destroyed RBCs, and iron in the blood that is transfused increases the amount of iron in the body and leads to severe and chronic iron overload. This is the main cause of damage to major organs the heart, liver, pancreas and kidneys especially.
In order to facilitate the excretion of excess iron in the body and rectify iron overload individuals need to have iron chelation therapy. This is done by self injection (in the case of children this is done by the parents) of a chelating agent called desferrioxamine (Desferal). This is administered using a continuous subcutaneous route via a chelation pump, and is given on average for five nights a week, running for approximately eight hours per therapy session.
The effectiveness of chelation is dependent on the individual’s willingness and ability to adhere to the prescribed treatment. After many years of having four weekly blood transfusions, five nights a week chelation and other adjunct therapy makes living with Thalassaemia major cumbersome and individuals may fail to maintain their treatment, thus risking development of fatal complications especially that of iron overload. As with other life long chronic conditions the psychological management of this patient group is essential and need to be part of a holistic care plan.
The oral chelator Deferiprone is not as effective in removing excess iron as desferrioxamine but is better than nothing for those patients who have difficulty complying with the use of subcutaneous chelation therapy. The oral chelator Exjade has been shown to be more effective than Deferiprone however its cost makes it somewhat prohibitive; whilst some health care purchasers (Primary Care Trusts in the NHS) have agreed to fund their use others are reluctant to give their approval but the drug has become more widely used in the some countries.
Other therapies for this patient group include daily Penicillin and use of vitamin C to aid Desferal in its role as a chelating agent. Un-transfused patients are also given folic acid 5mg / week.
Close monitoring of cardiac, liver and endocrine function are vital in order to identify impending complications with the intention of preventing them if possible or managing them if they cannot be fully controlled. Pubertal development need to be closely monitored and managed.